Type B Meetings
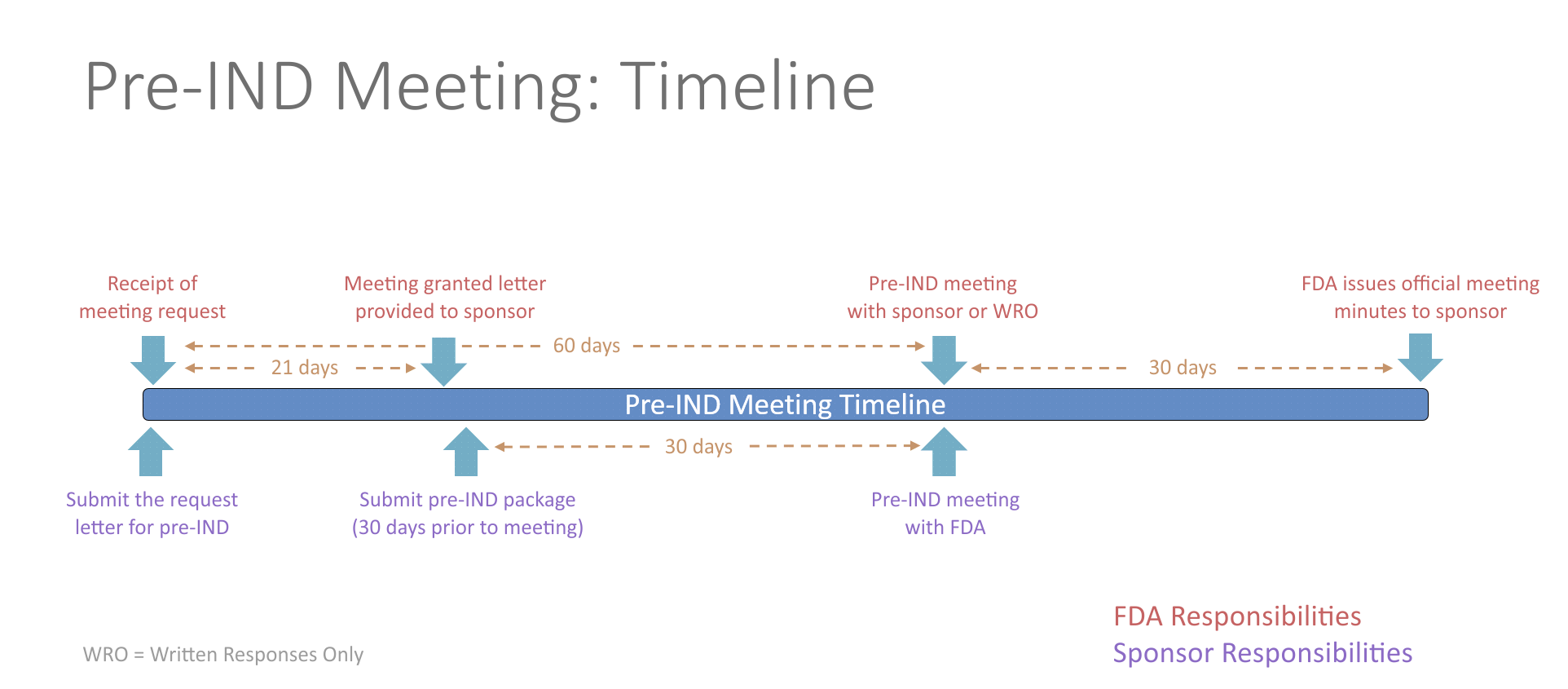
Pre-IND Meeting
The FDA allows for one pre-IND meeting prior to IND submission to discuss any questions or concerns concerning the clinical trial approach. Any topics may be discussed during this meeting including questions concerning general product development, manufacturing information, nonclinical testing, protocol design or other regulatory questions. Overall, meeting with the FDA has proven to decrease drug development time. There is no fee to the investigator associated with this meeting.
The process for requesting a pre-IND meeting starts by submitting a meeting request letter to the FDA, which includes a brief product description, proposed regulatory pathway, proposed indication(s) or context of product development, objectives and expected outcomes, proposed agenda, preliminary questions, sponsor attendees, proposed meeting format and dates for the meeting. Meetings last up to one hour via an in-person meeting or teleconference. Alternatively, sponsors can request written responses only in lieu of a meeting. The FDA will respond with the meeting date and the type of meeting granted. The study team will then need to send a briefing package 30 days prior to the meeting with finalized questions and information. The FDA will provide preliminary comments about 2 days before the meeting, providing you time to review their responses prior to your discussion with them. If the sponsor determines the preliminary comments are sufficient, the meeting can be cancelled. The meeting should be focused on any outstanding questions remaining. No new questions or proposals may be raised during the pre-IND meeting.
More information about pre-IND meetings
FDA guidance on Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products
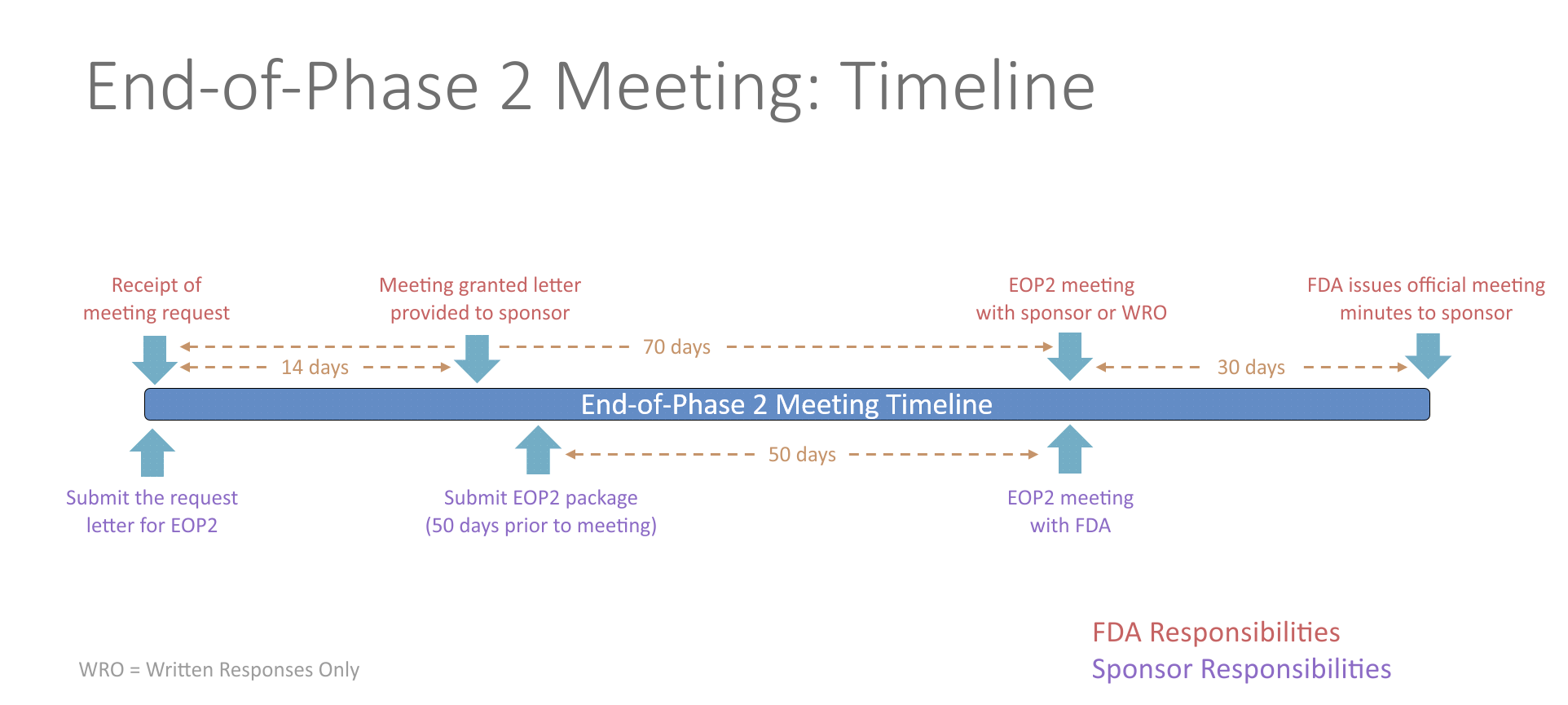
End-of-Phase 2 Meeting
An end-of-Phase 2 meeting (EOP2) is a formal meeting between the sponsor of an IND, the regulatory contact, and the FDA. The purpose of an EOP2 meeting is to determine the pathway for proceeding to a Phase 3 study, to evaluate the Phase 3 plan and protocol for adequacy, to assess pediatric safety and effectiveness, and to identify any additional information that would be needed to support a marketing application. This provides the sponsor an opportunity to get FDA input prior to conducting an expensive Phase 3 study.
The FDA will schedule the EOP2 meeting within 14 days of receiving the written meeting request. At least 50 days prior to the EOP2 meeting, the sponsor should submit a meeting package containing the plan for Phase 3, summaries of Phase 1 and 2 investigations, specific protocols for Phase 3 studies, plans for pediatric studies, and tentative labeling for the drug, if available. The initial meeting request and the meeting package should contain a proposed agenda and a list of questions. Any agreements reached during the meeting will be recorded in the FDA meeting minutes and provided to the sponsor. Studies conducted in accordance with the agreement will be presumed to be sufficient in objective and design for the purpose of obtaining marketing approval of the drug.
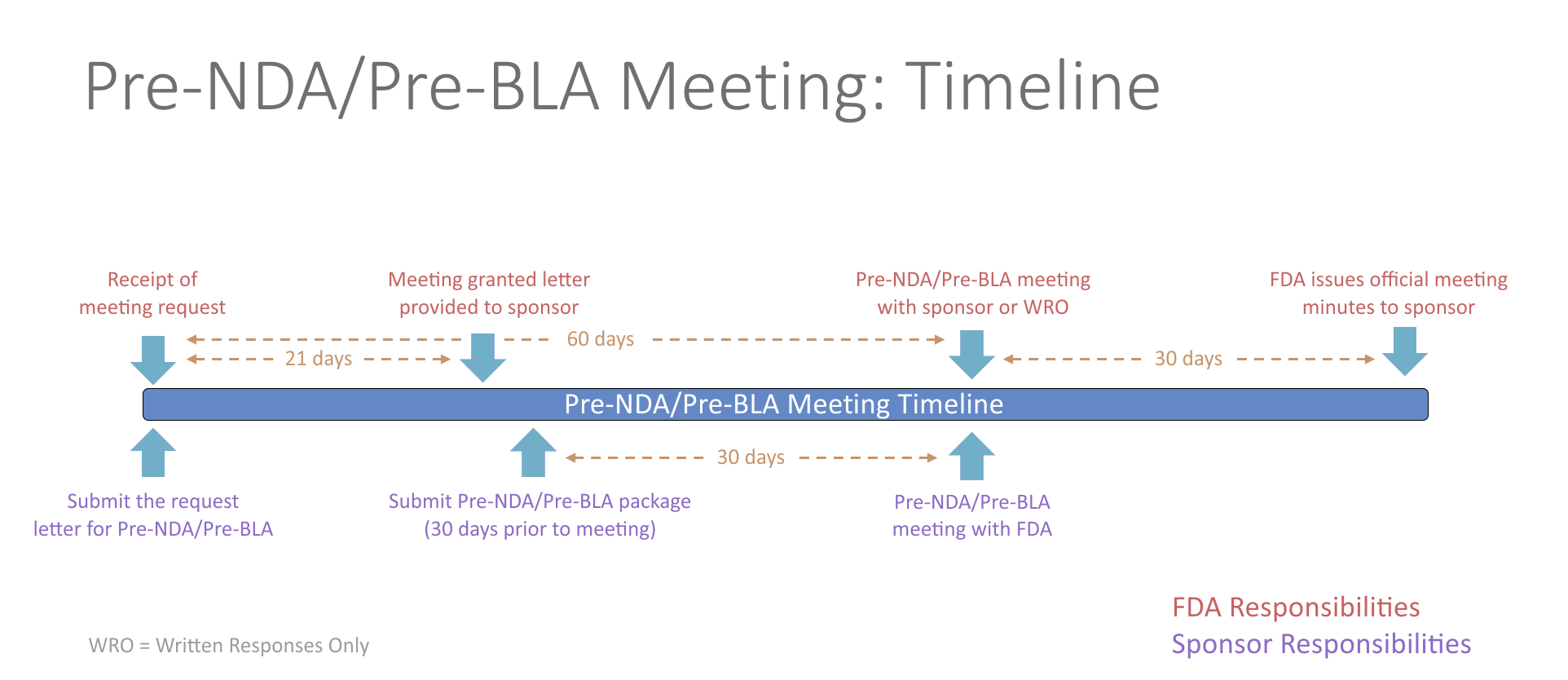
Pre-NDA/Pre-BLA Meetings
The pre-New Drug Application (NDA)/pre-Biologics License Application (BLA) meeting is a critical meeting between the sponsor and FDA to ensure the submission of a well-organized and readily reviewable NDA/BLA. This includes a discussion of the format and content of the anticipated application, and the presentation of data, the dataset structure, the acceptability of data for submission, and the projected submission date of the application. The meeting should be held in advance of the planned marketing application submission to allow for a meaningful response to FDA feedback and should generally occur no less than 60 days prior to the planned submission.
Potential topics for questions at the pre-NDA/pre-BLA phase of development include questions surrounding formatting of the submission, such as regulatory requirements, organization of the submission, the electronic common technical document (eCTD), and questions related to evidence of effectiveness seen in the Phase 3 study. The pre-NDA/pre-BLA meeting helps to determine whether outstanding issues require additional data or studies/trials that may affect the ability to file the future submission.
Examples of pre-NDA/pre-BLA meeting topics include: unresolved issues, trials to support quality, safety and efficacy, pediatric studies, data summary, data format and presentation, drug name review, new molecular entities (NMEs), preliminary discussions of risk management plans, post-marketing studies or trials, quality information and inspection considerations, outline of data to be submitted for abuse potential assessment and drug scheduling.