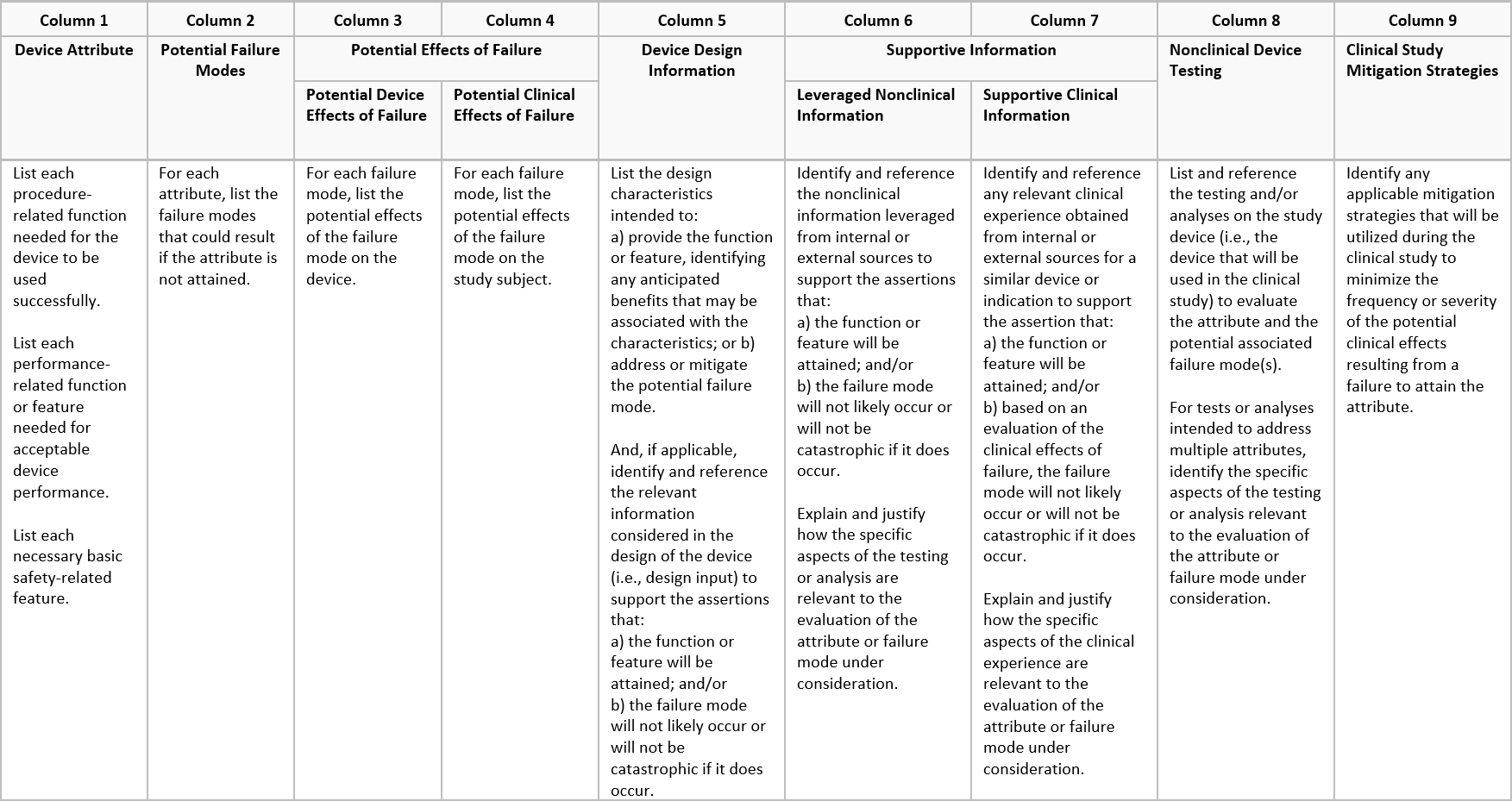
Strategy table example
Maintain all of the headings in this document. If some are not applicable, simply state this under the appropriate headings.
When the submission is finalized, be sure to save as a PDF and rename according to eCopy guidelines.
While the FDA has not officially outlined how they prefer the contents of an IDE submission to be presented, including the information listed in the sections below will provide the FDA with the information necessary to conduct a successful review.
Although a manufacturer may be supplying the device being investigated, the FDA defines the study "sponsor" as the person, company, or institution who initiates the study. The sponsor is also responsible for the regulatory aspects of the study. Often in an academic setting the investigator is called a "sponsor-investigator" because they assume both roles of the research.
Provide the following information in this section:
It is also strongly encouraged to also provide information for a designated alternate contact who is authorized by the sponsor to correspond with the FDA in the event that the sponsor cannot be reached. This person is often the project manager or regulatory coordinator for the investigation.
In this section, the sponsor should provide a complete report of prior investigations of the device.
The report of prior investigations shall include reports of all prior clinical, animal, and laboratory testing of the device and shall be comprehensive and adequate to justify the proposed investigation.
A bibliography of all publications, whether adverse or supportive, that are relevant to an evaluation of the safety or effectiveness of the device, copies of all published and unpublished adverse information, and, if requested by an IRB or FDA, copies of other significant publications.
A summary of all other unpublished information (whether adverse or supportive) in the possession of, or reasonably obtainable by, the sponsor that is relevant to an evaluation of safety or effectiveness of the device.
If information on nonclinical laboratory studies is provided, a statement that all such studies have been conducted in compliance with applicable requirements in the good laboratory practice (GLP) regulations in 21 CFR part 58. If the study was not conducted in compliance with such regulations, a brief statement of the reason for the non-compliance.
At the beginning of this section, the sponsor can give a brief overview of the investigational plan, including the rationale for the trial, whether it is a single or multi-site study, and a description of the endpoints.
The name and intended use of the device and the objectives and duration of the investigation.
A written protocol describing the methodology to be used and an analysis of the protocol demonstrating that the investigation is scientifically sound.
The NIH-FDA Protocol Template is a highly recommended template that guides investigators through the information that should be included in a protocol.
A description and analysis of all increased risks to which subject will be exposed by the investigation; the manner in which these risk will be minimized; a justification that the risks are reasonable in relation to the expected benefits; and a description of the patient population including the number, age, sex, and condition.
The risk analysis should include the anticipated benefits and potential clinical effects of failure identified in the device evaluation strategy, as well as risks independent of the device that may be related to the underlying disease comorbidities, or inherent to the procedure, and benefits unique to the device concept. For example, a risk analysis may include the risks associated with use of anesthetic and contrast agents and the benefits of a less invasive intervention.
Methods to minimize risks may include the use of standard approaches, with additional mitigation strategies to protect individual study subjects and future study participants during the ongoing study. Examples of both standard and additional risk mitigation strategies include:
use of study sites that have sufficient expertise and resources to manage adverse events and provide appropriate alternative therapies if needed;
identification of qualified investigators with adequate training to conduct the early feasibility study;
a plan to capture human factors information during the course of the study to modify the procedures or device as necessary based on the information obtained;
specifying appropriate study inclusion and exclusion criteria;
limiting the sample size to a reasonable number for an early feasibility study (e.g., 5-10 initial subjects);
follow-up assessments at regular intervals to monitor subject safety and device effectiveness (i.e., potentially more frequent than for a traditional feasibility or pivotal study);
timely reporting of serious adverse events (e.g., after each occurrence rather than only in a periodic progress report);
timely reporting of device performance parameters, which help determine whether the device functions as intended (e.g., measurements of deliverability, stability, handling, visualization, patency, integrity);
non-sequential enrollment, that is, initial device use in subjects with more favorable anatomical characteristics as compared to the population otherwise eligible for the early feasibility study (e.g., selecting subjects that meet study eligibility requirements but do not have anatomic features that may increase the difficulty of device use); and
a pre-specified plan for periodic patient outcome assessments and reporting prior to enrollment of additional patients (e.g., as frequently as after each use of the device).
One way to present the risk/benefit assessment is through a device evaluation strategy table.
The process of constructing the device evaluation strategy table can be divided into four parts:
A description of each important component, ingredient, property and principle of operation of the device and of each anticipated change in the device during the course of investigation. Include any drawings, photos, videos, or design information.
Make sure your device description is clear and describes ALL elements of your proposed device (e.g., physical description, figures, materials of construction, software documentation), in addition to providing its dimensions and all of the dimensions of its components. Diagrams of both your device and an exploded view of your device with all of the components identified are very helpful.
Clearly identify how all components of your device fit together and are held together.
Clearly describe the functional purpose of each element of your device. This helps FDA both understand the components of your device and your device as a whole.
Clearly identify all of the different sizes and configurations your device comes in. It is often helpful if this is done in a tabular format.
Always make sure you use consistent terminology for each component of your device in your submission.
The sponsor's written procedures for monitoring the investigation and the name and address of each monitor.
The following language can be used for studies conducted at a single site where only one investigator, the sponsor-investigator, is involved in the study:
"A risk-based monitoring plan will be developed and implemented for the proposed clinical study.
Based on the latest FDA guidance on IDE policies and procedures (January 20, 1998), the submission of written monitoring procedures is not required for a sponsor-investigator initiated study where only one investigator will be involved in the study. The sponsor-investigator, Dr. [insert name], will serve as the study monitor for the clinical study proposed under this IDE."
If you are using a marketed device, then it is appropriate to refer to the product label and provide copy or a URL to the most current product label. If any modifications have been made, provide details on all changes.
If you have a Letter of Authorization (LoA) from another sponsor referencing their FDA submission (IND, NDA, BLA, IDE, DMF, etc), include the LoA in this section. The LoA serves the purpose to allow the FDA reviewer to review their submission on file in relation to your IDE application.
If you are manufacturing the device, include a description of the methods, facilities, and controls used for the manufacture, processing, packing, storage, and, where appropriate, installation of the device, in sufficient details so that a person generally familiar with good manufacturing practice can make a knowledgeable judgment about the quality control used in the manufacture of the device.
An example of the agreement to be entered into by all investigators to comply with investigator obligations under this part, and a list of the names and addresses of all investigators who have signed the agreement. Information that must be included in the written agreement can be found in §812.43 .
The investigators agreement must include:
The investigator's CV;
Where applicable, a statement of the investigator's relevant experience (including the dates, location, extent and type of experience);
If the investigator was involved in an investigation or other research that was terminated, an explanation of the circumstances that led to termination;
Investigator's commitment to provide sufficient and accurate financial disclosure information and update information if any relevant changes occur during the investigation and for one year following the completion of the study.
A statement of the investigator's commitment to:
Conduct the investigation in accordance with the agreement, the investigational plan, Part 812 and other applicable FDA regulations, and conditions of approval imposed by the reviewing IRB and FDA;
Supervise all testing of the device involving human subjects;
Ensure that the requirements for obtaining informed consent are met
A statement that all investigators who will participate in the investigation have signed the agreement, that the list of investigators includes all the investigators participating in the investigation, and that no investigator will be added to the investigation until they have signed the agreement.
The following statement can be used to satisfy this requirement: "As required for an IDE study, we commit to obtain a signed investigator agreement from all current investigators who are participating in the investigation. Additionally, no future investigators will be added until they have signed the agreement."
A list of the name, address, and chairperson of each IRB that has been or will be asked to review the investigation and a certification of the action concerning the investigation taken by such IRB.
The name and address of any institution at which a part of the investigation may be conducted.
If the device is to be sold, the amount to be charged and an explanation of why sale does not constitute commercialization of the device.
A claim for categorical exclusion under § 25.30 or § 25.34 or environmental assessment under § 25.40.
Include the following statement: "Please note that an environmental assessment as required under 21 CFR 25.40 or a claim for categorical exclusion under 21 CFR 25.30 or 25.34 is no longer required [§25.34(g)]."
Include copies of all labeling for the device. (If you are using a marketed device, then it is appropriate here to refer to the most current product labeling and provide a URL link to the most current labeling.)
Labeling is defined as "all labels and other written, printed, or graphic matter upon any article or any of its containers or wrappers, or accompanying such article at any time while a device is held for sale after shipment or delivery for shipment in interstate commerce."
An investigational device or its immediate package must bear a label with the following information:
The name and place of business of the manufacturer, packer, or distributor;
The quantity of contents, if appropriate; and
The statement, "CAUTION Investigational device. Limited by Federal (or United States) law to investigational use."
The label must also describe all relevant contraindications, hazards, adverse effects, interfering substances or devices, warnings, and precautions.
The labeling of an investigational device must not contain any false or misleading statements nor imply that the device is safe or effective for the purposes being investigated.
Copies of all forms and informational materials to be provided to subjects to obtain informed consent. Information that should be included in informed consent documents can be found in §50.25(a) .
A statement that the study involves research
An explanation of the purposes of the research
The expected duration of the subject's participation
A description of the procedures to be followed
Identification of any procedures which are experimental (i.e., not standard of care)
A description of any reasonably foreseeable risks or discomforts to the subject (ideally subdivided by frequency and severity)
A description of any benefits to the subject or to others which may reasonably be expected from the research
A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject
A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained
For research involving more than minimal risk, an explanation as to whether any compensation, and an explanation as to whether any medical treatments are available, if injury occurs and, if so, what they consist of, or where further information may be obtained
An explanation of whom to contact for answers to pertinent questions about research subjects' rights, about the research and in the event of research-related injury.
A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits, to which the subject is otherwise entitled
Include clinicaltrials.gov language
FDA-regulated clinical investigations: Subjects must be informed that the FDA may inspect the records of the study.
A statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant), which are currently unforeseeable
Anticipated circumstances under which the subject's participation may be terminated by the investigator without regard to the subject's consent
Any additional costs to the subject that may result from participation in the research
The consequences of a subject's decision to withdraw from the research and procedures for orderly termination of participation by the subject
Statement that significant new findings developed during the course of the research, which may relate to the subject's willingness to continue participation, will be provided to the subject
The approximate number of subjects involved in the study
A clear description of the operation of the bio-specimen resource. This description could include details that may be of interest to human research participants, such as whether identifiable information will be maintained by the bio-specimen resource and/or whether research results will be linked to the bio-specimen.
The conditions under which samples and data will be released to recipient investigators. Procedures for protecting the privacy of human research participants and confidentiality of data.
Specific descriptions of the nature and purpose of the research.
Information about the consequences of DNA typing if human genetic research is anticipated.
Specific and meaningful description of what will be used or disclosed.
The name or other specific identification of the person, or class of persons, authorized to make the use or disclosure.
The name or other specific identification of the person, or class of persons, to whom the covered entity may make the requested use or disclosure.
A description of each purpose of the requested use or disclosure.
An expiration date/expiration event that relates to the individual or the purpose of the use or disclosure.
Statement regarding the right of the individual to revoke the authorization in writing, and the limits of that right.
Statement regarding the right of the individual to refuse to sign authorization
Statement regarding ability or inability to condition treatment, payment, enrollment or eligibility for benefits on the authorization
Re-disclosure Statement - Information disclosed to others not subject to the Privacy Rule may be re-disclosed by them without the Privacy Rule protections (cannot promise that information will definitely be protected)
Any other relevant information FDA requests for review of the application.
This is a good place to list any references that will be attached to the application.
Additionally, a complete IDE template can be downloaded:
One eCopy and one paper cover letter:
One original hard copy of the cover letter
Include a handwritten or valid digital signature
Include the submission tracking number, if previously assigned
Use the company letterhead and include full contact information
Provide a brief description of the purpose of the submission along with submission type (i.e. IDE) and stage of review (i.e. original, amendment, supplement, or report)
One electronic copy (eCopy) of the submission on digital media
All documents should be in Portable Document Format (PDF)
Individual PDFs must be 50MB or smaller in size
Remove any password protections
No embedded attachments or attributes
If non-PDFs are required, zip all non-PDF content into one file and save within a folder labeled either "STATISTICAL DATA" or "MISC FILES"
Follow the eCopy PDF naming convention described in the FDA eCopy guidance (see link below)
It is recommended that the cover letter also be included as a PDF in the eCopy, but it is not required.
An electronic copy (eCopy) is an electronic version of your medical device submission stored on a compact disc (CD), digital video disc (DVD), or a flash drive. Including an eCopy with your submission has been required since January 1, 2013, and a final rule was issued by FDA on December 13, 2019 requiring medical device premarket submissions to be sent in electronic format, eliminating the need for paper submissions. A submission with an eCopy that does not meet the technical standards outlined in the eCopy guidance will be placed on eCopy hold until a valid eCopy is received.
The following resources will help you in understanding the eCopy program and how to successfully create and submit your eCopy:
eSubmitter-eCopies Tool - a voluntary tool that formats your eCopy content and allows you to download onto a local drive
eCopies Validation Module - a voluntary tool that verifies the format of an eCopy you have already developed on your local drive
If you have additional questions about the eCopy program, please contact the eCopy Program Coordinators at CDRH-eCopyinfo@fda.hhs.gov or 240-402-3717.
These are current addresses, but please confirm on the FDA website:
For devices regulated by the Center for Devices and Radiological Health (CDRH):
For devices regulated by the Center for Biologics Evaluation and Research (CBER):
You must state on the outer packaging (e.g. The FedEx label) of each submission what the submission contains. For example, an "IDE application", a "supplemental IDE application" or a "correspondence concerning an IDE application". This should also be clearly stated on your cover letter in the "RE:" section.
For submissions to CDRH, the initial submission is usually sent to the attention of the appropriate Division Director if you know where the subject device or similar devices are reviewed. For CBER submissions, the addressee may be the appropriate Office Director or Regulatory Project Manager where the subject device or similar devices are reviewed.
The CDRH Management Directory on the FDA website can be helpful in identifying the appropriate review division or Division Director to be addressed.
The CDRH Office of Product Evaluation and Quality (OPEQ) website may also be helpful in identifying the appropriate review division.
For CBER submissions, the CBER Key Staff Directory may be helpful in identifying the appropriate Office Director or Regulatory Project Manager.