Frequently Asked Questions
Drug/Biologic Questions
- What is a Pre-IND meeting?
- Are there other milestones when Investigators should meet with the FDA?
- Can I submit my IRB application before I know if I need an IND?
- What is GMP?
Device Questions
Other Regulatory Questions
- What is GLP?
- What is GCP?
- What is clinicaltrials.gov, and do I need to register my study?
- What are the additional clinical research protections for children?
Drug/Biologic Questions
What is a Pre-IND meeting?
- The FDA allows for one pre-IND meeting prior to IND submission to discuss any questions or concerns concerning the clinical trial approach. Any topics may be discussed during this meeting including questions concerning general product development, manufacturing information, nonclinical testing, protocol design or other regulatory questions. Overall, meeting with the FDA has proven to decrease drug development time. There is no fee to the investigator associated with this meeting.
- The process for requesting a pre-IND meeting starts by submitting a meeting request letter to the FDA, which includes a brief product description, proposed regulatory pathway, proposed indication(s) or context of product development, objectives and expected outcomes, proposed agenda, preliminary questions, sponsor attendees, proposed meeting format and dates for the meeting. Meetings last up to one hour via an in-person meeting or teleconference. Alternatively, sponsors can request written responses only in lieu of a meeting. The FDA will respond with the meeting date and the type of meeting granted. The study team will then need to send a briefing package 30 days prior to the meeting with finalized questions and information. The FDA will provide preliminary comments about 2 days before the meeting, providing you time to review their responses prior to your discussion with them. If the sponsor determines the preliminary comments are sufficient, the meeting can be cancelled. The meeting should be focused on any outstanding questions remaining. No new questions or proposals may be raised during the pre-IND meeting.
-
More information about pre-IND meetings
FDA page on Pre-IND meetings
FDA guidance on Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products 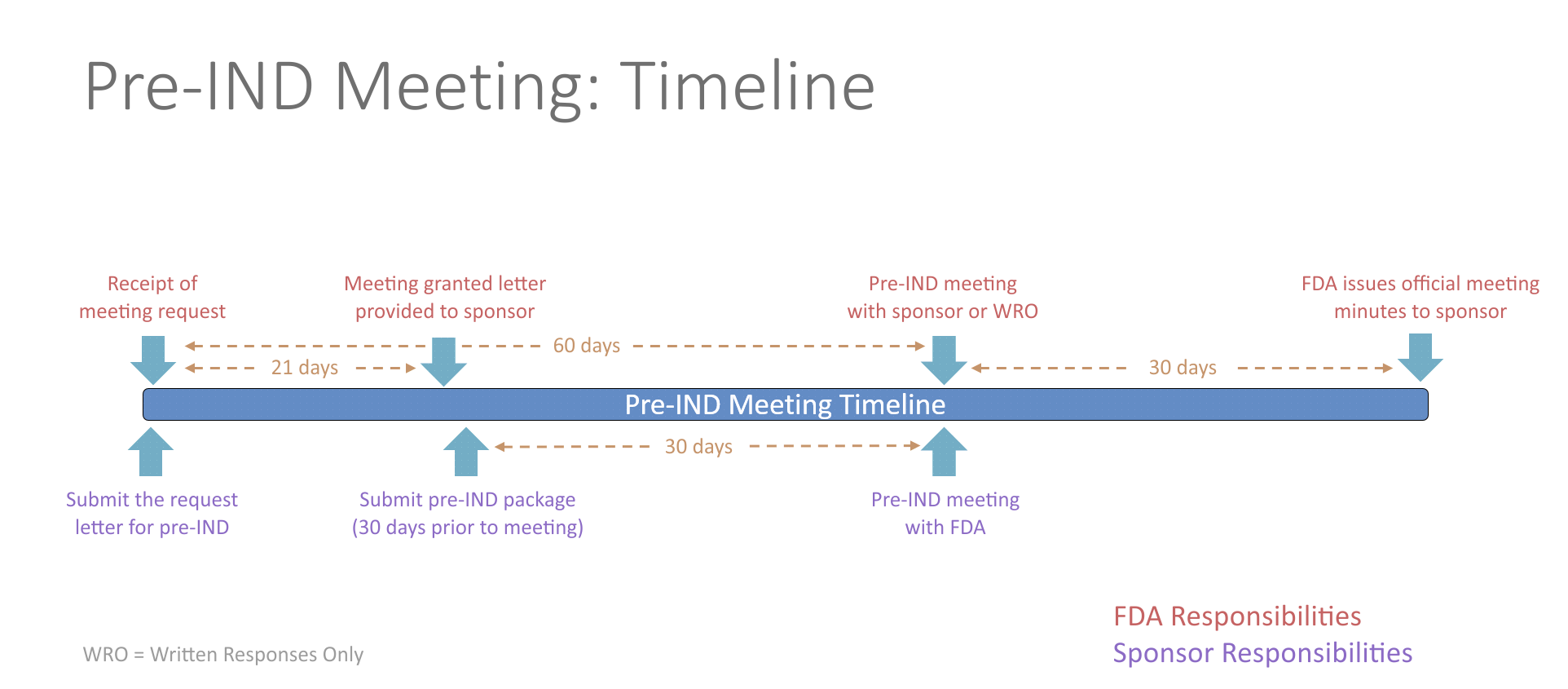
Are there other milestones when Investigators should meet with the FDA?
- Yes! They are itemized below. For additional information on meetings with FDA, please see the FDA guidance on Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products .
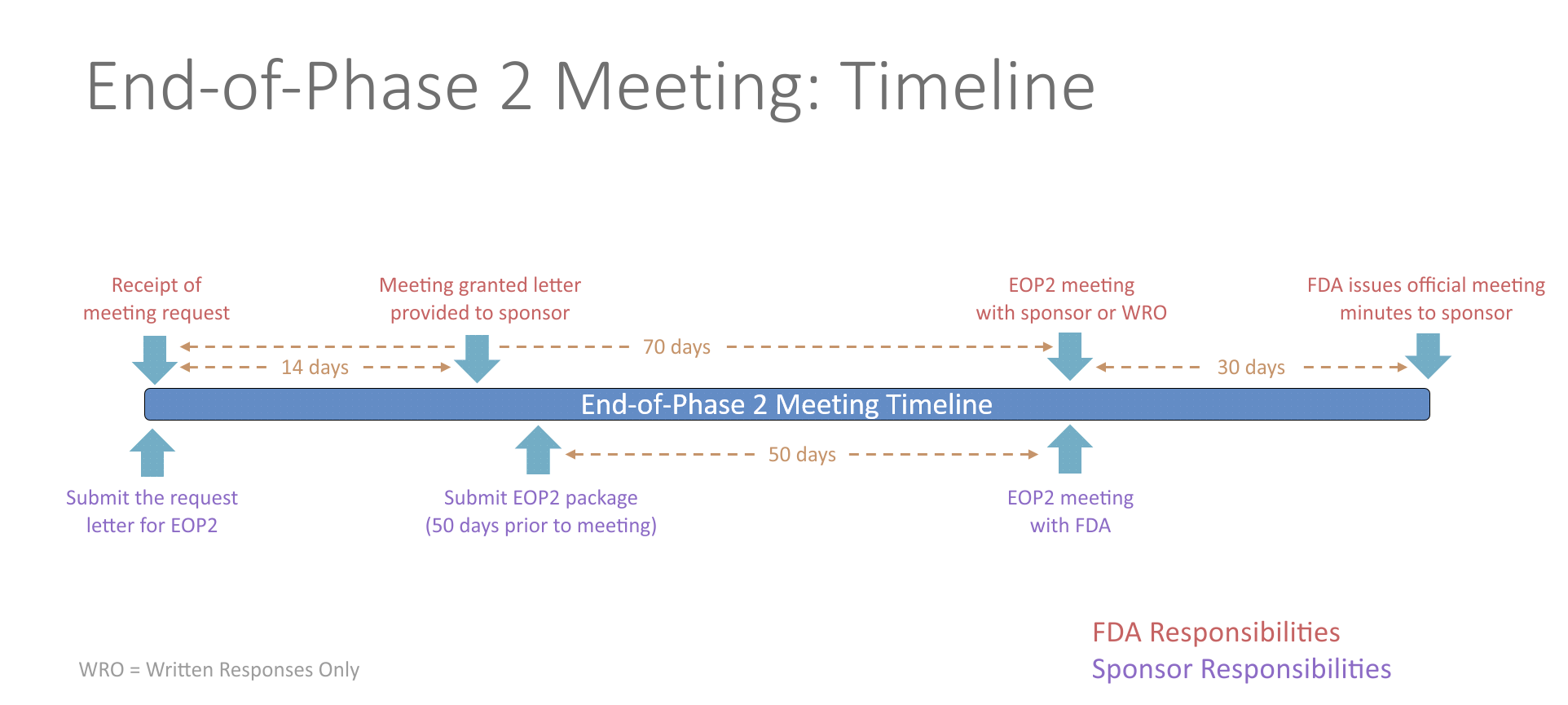
- End-of-Phase 2 Meeting
- An end-of-Phase 2 meeting (EOP2) is a formal meeting between the sponsor of an IND, the regulatory contact, and the FDA. The purpose of an EOP2 meeting is to determine the pathway for proceeding to a Phase 3 study, to evaluate the Phase 3 plan and protocol for adequacy, to assess pediatric safety and effectiveness, and to identify any additional information that would be needed to support a marketing application. This provides the sponsor an opportunity to get FDA input prior to conducting an expensive Phase 3 study.
- The FDA will schedule the EOP2 meeting within 14 days of receiving the written meeting request. At least 50 days prior to the EOP2 meeting, the sponsor should submit a meeting package containing the plan for Phase 3, summaries of Phase 1 and 2 investigations, specific protocols for Phase 3 studies, plans for pediatric studies, and tentative labeling for the drug, if available. The initial meeting request and the meeting package should contain a proposed agenda and a list of questions. Any agreements reached during the meeting will be recorded in the FDA meeting minutes and provided to the sponsor. Studies conducted in accordance with the agreement will be presumed to be sufficient in objective and design for the purpose of obtaining marketing approval of the drug.
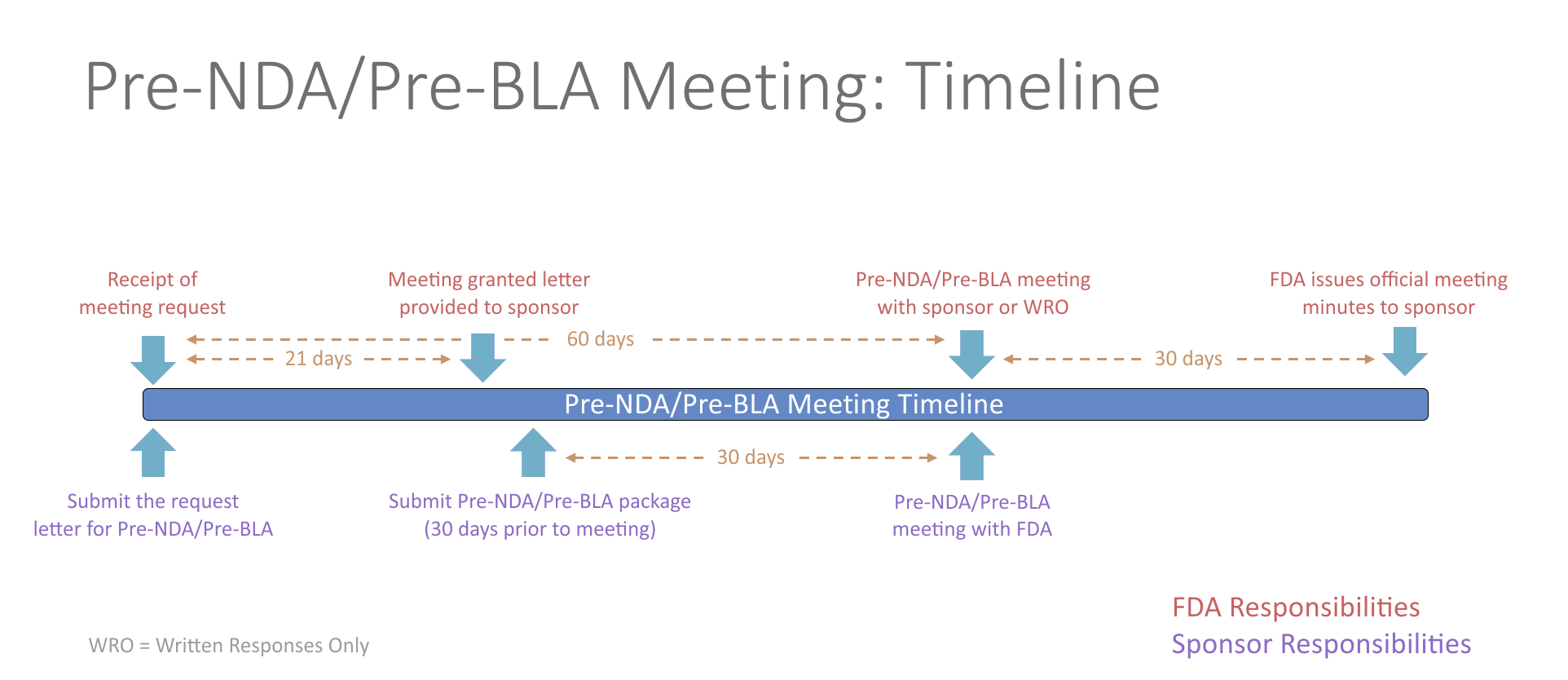
- Pre-NDA/Pre-BLA Meetings
- The pre-New Drug Application (NDA)/pre-Biologics License Application (BLA) meeting is a critical meeting between the sponsor and FDA to ensure the submission of a well-organized and readily reviewable NDA/BLA. This includes a discussion of the format and content of the anticipated application, and the presentation of data, the dataset structure, the acceptability of data for submission, and the projected submission date of the application. The meeting should be held in advance of the planned marketing application submission to allow for a meaningful response to FDA feedback and should generally occur no less than 60 days prior to the planned submission.
- Potential topics for questions at the pre-NDA/pre-BLA phase of development include questions surrounding formatting of the submission, such as regulatory requirements, organization of the submission, the electronic common technical document (eCTD), and questions related to evidence of effectiveness seen in the Phase 3 study. The pre-NDA/pre-BLA meeting helps to determine whether outstanding issues require additional data or studies/trials that may affect the ability to file the future submission.
- Examples of pre-NDA/pre-BLA meeting topics include: unresolved issues, trials to support quality, safety and efficacy, pediatric studies, data summary, data format and presentation, drug name review, new molecular entities (NMEs), preliminary discussions of risk management plans, post-marketing studies or trials, quality information and inspection considerations, outline of data to be submitted for abuse potential assessment and drug scheduling.
- The FDA training course titled "Engaging with the FDA During New Drug Development" , is a useful course for those with novel drugs who need to request more information from the FDA regarding their current research path.
Can I submit my IRB application before I know if I need an IND?
- Yes, it is possible to submit an IRB application for a clinical study before an IND submission; however, if the IRB determines an IND may be needed, the study may not proceed until confirmation of IND exemption or acknowledgement of IND receipt is obtained from FDA and the 30 day review period has passed.
What is GMP?
- Current Good Manufacturing Practices (CGMP) are the regulatory requirements described in section 501(a)(2)(B) of the Federal Food, Drug, and Cosmetic Act (FD&C Act)(21 U.S.C. 351 (a)(2)(B)), which state the manufacturing conditions that must exist for a drug product manufacturer to supply drug that is not considered "adulterated" (21 CFR parts 210 and 211)
- The CGMP regulations for drugs contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a drug product. The CGMP regulations are in place to assure product safety and quality and that the product has the ingredients, purity, and strength it claims to have.
- The approval process for new drug and generic drug marketing applications includes a review of the manufacturer's compliance with the CGMP. FDA inspectors determine whether the firm has the necessary facilities, equipment, and skills to manufacture the new drug for which it has applied for approval. Decisions regarding compliance with CGMP regulations are based upon inspection of the facilities, sample analyses, and compliance history of the firm. This information is summarized in reports which represent several years of history of the firms.
- FDA understands that the information that is available on the manufacture of investigational products, especially Phase I investigational products may be limited; however, consistent with the FD&C Act (§ 501(a) (2) (B)), CGMP must be in effect for the manufacture of each batch of investigational drug used during phase 1 clinical trials. FDA has established specific guidance on the CGMP controls that should be in place during phase 1 clinical trials that are based on identified hazards for the manufacturing setting that follow good scientific and quality control principles. As the product progresses from Phase 1 through later phases of clinical development and more information is gained on the manufacturing process and controls, the FDA expects compliance with full CGMP requirements to increase and complete compliance with CGMPs should be in place by the time the product is marketed.
- For additional information on applying CGMPs in the manufacture of investigational new drugs used in phase 1 clinical trials, please see the FDA guidance on Current Good Manufacturing Practice for Phase 1 Investigational Drugs .
- Video explanation
Quality Assurance for IND & IDE studies Video explanation
Manufacturing for IND & IDE studies
Device Questions
What are general controls and special controls
- General controls are the basic provisions (authorities) of the Medical Device Amendments that provide the FDA with the means of regulating devices to ensure their safety and effectiveness. The general controls apply to all three classes of medical devices. They include provisions that relate to adulteration; misbranding; device registration and listing; premarket notification; banned devices; notification, including repair, replacement, or refund; records and reports; restricted devices; and good manufacturing practices.
- Special controls are regulatory requirements for class II devices. Special controls are usually device specific and include performance standards, post market surveillance, patient registries, special labeling requirements, and premarket data requirements.
- Exempt Devices
- Most Class I and a few Class II devices are exempt from the 510(k) premarket notification requirement because the FDA has determined that a 510(k) is not required to provide reasonable assurance of safety and effectiveness for the device. 510(k) exempt devices are still subject to other regulatory controls.
-
Exempt Device Examples
- Acupuncture needle
- Cardiopulmonary resuscitation (cpr) aid
- Catheter guide wire
- Conventional Urology Endoscopes
- Daily Wear Contact Lenses
- Dental Filling Materials
- Dental floss
- Dental syringe
- Ear, nose, and throat examination and treatment unit
- Electroencephalography
- Epistaxis balloon
- General Urological Catheters
- Hemorrhoidal ligator
- Ice bag
- Jaundice Monitors for Infants
- Lice removal kit
- liquid bandage
- Master hearing aid
- Medical scissors
- Menstrual Pads & Tampons (Cotton or Rayon, only)
- Nonrebreathing mask
- Pediatric position holder
- Pneumatic tourniquet
- Removable skin staple
- Surgical apparel
- Tongue depressor
- Tracheal tube fixation device
- Urethrotome
- Water vapor analyzer
- Wound Dressings
What is premarket approval (PMA)?
- Premarket approval (PMA) is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices. Class III devices are those that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury.
What is a 510(k)?
- A 510(k) is a premarket submission made to FDA to demonstrate that the device to be marketed is at least as safe and effective, that is, substantially equivalent, to a legally marketed device that is not subject to PMA. Submitters must compare their device to one or more similar legally marketed devices and support their substantial equivalency claims.
Other Regulatory Questions
What is GLP?
- In 1979, Good Laboratory Practice (GLP) regulations became effective under 21 Code of Federal Regulations (CFR) Part 58 which apply to all nonclinical safety studies intended to support research permits or marketing authorizations for products regulated by the US Food and Drug Administration (FDA), including food and color additives, animal food additives, human and animal drugs, medical devices for human use, biological products, and electronic products.
- Good laboratory practice (GLP) is a standard by which laboratory studies are designed, implemented, and reported to assure the public that the results are correct and the experiment can be reproduced exactly, at any time in the future.
What is GCP?
- Good Clinical Practice (GCP) is an international set of ethical and scientific quality standards for designing, conducting, recording and reporting human clinical trials to ensure the integrity of clinical research and protect the rights, safety, and well-being of trial subjects. The International Council on Harmonization (ICH) GCP guidance was developed in a collaborative effort with the World Health Organization (WHO) and regulatory agencies throughout the world.
- The US Food and Drug Administration (FDA) defines GCP as:
"A standard for the design, conduct, performance, monitoring, auditing, recording, analyses and reporting of clinical trials that provides assurance that the data and reported results are credible and accurate, and that the rights, integrity, and confidentiality of trial subjects are protected."
- The US FDA has collaborated and adopted the ICH GCP guidance for the conduct of clinical trials, and FDA's GCP guidance document can be found here . GCP is applied when conducting trials with human subjects, and should be followed when generating clinical trial data that are intended to be submitted to regulatory authorities.
What is clinicaltrials.gov, and do I need to register my study?
- Clinicaltrials.gov is a registry of clinical trials operated by the National Institutes of Health (NIH). Applicable clinical studies involving drugs, biologics, and devices must be registered on the site. Additionally, any clinical trial funded in whole or in part by NIH, even if it does not involve a drug, biologic, or device must be registered. In 2016, legislation titled the "Final Rule" introduced new reporting requirements for clinical trials, including the mandatory reporting of results for applicable studies. For more information on clinicaltrials.gov and the Final Rule, please visit Submit Studies and Support Materials .
- To learn more about clinicaltrials.gov and your responsibilities as a sponsor-investigator, see the following videos:
- Video explanation
All clinicaltrials.gov videos
What are the additional clinical research protections for children?
- Due to the vulnerable nature of infants and children, the FDA has put additional regulations in place to ensure the safety and wellbeing of underage clinical research subjects. Modified informed consent practices, and additional documentation in IND/IDE submissions are among the requirements necessary to conduct clinical investigations in children.
- For more information, please visit FDA's Additional Protections for Children page.