Is My Study Exempt?
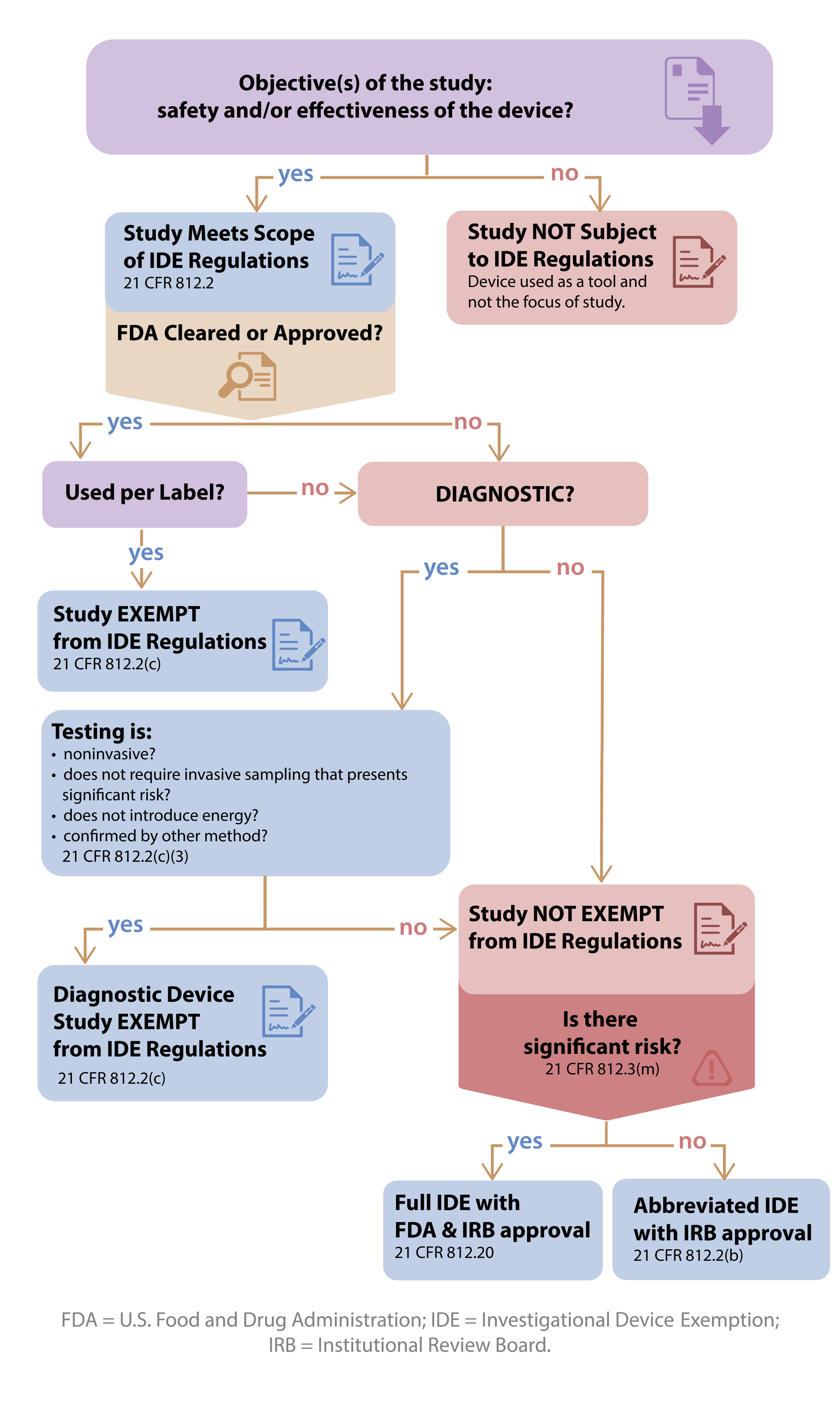
Please see FDA guidance for information on determining significant vs non-significant risk.
What Is a Medical Device?
The definition of the term device in section 513(a)(1) of the FD&C Act is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent or other similar or related article or component part or accessory which:
- is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease
- is intended to affect the structure or any function of the body
- does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes
Non-traditional medical devices
In thinking about medical devices, some obvious examples may come to mind, like a pacemaker, an MRI, or a blood pressure cuff. However, there are some other "less traditional" devices that also require FDA oversight as a medical device. They are described below.
In vitro diagnostics (IVDs)
- IVDs are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions.
- For example, pregnancy test, HIV blood screening test, oncogene testing
- Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
- ex. blood, spinal fluid, tissue samples, serum, urine
- The FDA considers the IVD to be the entire process from specimen collection to results reporting:
- Specimen collection and transport
- Specimen preparation
- Specimen examination/analysis
- Method of calculating/reporting result
- Clinical studies, particularly drug studies, often contain objectives with the purpose of determining whether biomarkers correlate with disease state, treatment response, or risk of disease. Depending on study design, this analysis can be considered the early development of an IVD that is subject to 21 CFR 812 .
- Some things to consider when deciding if 21 CFR 812 may apply are whether the targets for the analysis are specific and well-defined, how much is previously known about the biomarkers, and/or how the disease correlating signature is used.
Software Functions and Mobile Medical Applications
- FDA has regulatory oversight of software functions that meet the definition of a device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), and these software functions are referred to as "device software functions." Device software functions may include "Software as a Medical Device (SaMD)" and "Software in a Medical Device (SiMD)."
- A Mobile Medical Application (MMA) ". . .is a mobile app that incorporates device software functionality that meets the definition of device in section 201(h) of the FD&C Act; and either is intended:
- to be used as an accessory to a regulated medical device; or
- to transform a mobile platform into a regulated medical device."
- Device software functions may be used on a mobile platform (e.g., mobile medical apps), other general-purpose computing platform, or in the function or control of a hardware device.
- Generally, if a software function is intended to perform a medical device function (i.e. to diagnose disease or other conditions, or cure, mitigate, treat, or prevent disease), it is regulated as a medical device, regardless of the platform on which it is run.
- FDA intends to focus its regulatory oversight only to those device software functions that could pose a risk to a patient’s safety if they fail to function as intended:
- "Software functions that are an extension of one or more medical devices by connecting to such device(s) for purposes of controlling the device(s) or analyzing medical device data."
- For example, software that provides the ability to control inflation and deflation of a blood pressure cuff through a mobile platform
- "Software functions (typically, mobile apps) that transform the mobile platform into a regulated medical device by using attachments, display screens, or sensors or by including functionalities similar to those of currently regulated medical devices."
- For example, a software function that uses the built-in accelerometer on a mobile platform to collect motion information for monitoring sleep apnea
- "Software functions that become a regulated medical device by performing patient-specific analysis and providing patient-specific diagnosis, or treatment recommendations."
- For example, software functions that use patient-specific parameters and calculate dosage or create a dosage plan for radiation therapy
- "Software functions that are an extension of one or more medical devices by connecting to such device(s) for purposes of controlling the device(s) or analyzing medical device data."
- For software functions that fall under FDA regulatory oversight, the IDE regulations are enforced and apply when the objective of the clinical investigation is to assess the safety and/or effectiveness of the software function.
- FDA intends to exercise enforcement discretion (meaning it does not intend to enforce requirements under the FD&C Act) for software functions that:
- "Help patients (i.e., users) self-manage their disease or conditions without providing specific treatment or treatment suggestions; or
- Automate simple tasks for health care providers."
- For software functions that fall under FDA enforcement discretion, the IDE regulations are not currently being enforced.
Additional information on software and mobile medical apps, and more examples of software that fall into the various regulatory oversight categories, can be found in the FDA Guidance for Industry and FDA Staff - Policy for Device Software Functions and Mobile Medical Applications .
IDE Exemption Criteria
When is a clinical investigation a "device study"?
- If the objective of the clinical investigation is to assess the safety and/or effectiveness of a medical device, then the study is a device study and is subject to regulatory oversight by the US Food and Drug Administration as defined in 21 CFR 812 (Investigational Device Exemption).
- If the objective of the study is not to test the safety or effectiveness of the device, then the study would not fall within the scope of 21 CFR 812 .
- Such a device is used as a "tool". One example of this would be when an MRI is used to collect data in an oncology drug trial to evaluate tumor response or a thermometer used to check temperature as an inclusion criteria for a study.
When is a device study exempt from IDE regulations?
A device investigation is exempted from the IDE regulations if the device fits any of the following criteria (21 CFR 812.2(c) ):
- A legally marketed device when used in accordance with its labeling.
- This criterion applies to devices with an approved PMA or 510(k) clearance, but does not apply to transitional devices, which are devices that were regulated by FDA as new drugs before May 28, 1976
- A diagnostic device if it complies with the labeling requirements in §809.10(c) and if the testing (see below for more details):
- is noninvasive;
- does not require an invasive sampling procedure that presents significant risk;
- does not by design or intention introduce energy into a subject; and
- is not used as a diagnostic procedure without confirmation by another medically established diagnostic product or procedure;
- A device undergoing consumer preference testing, testing of a modification, or testing of a combination of two or more devices in commercial distribution, if the testing is not for the purpose of determining safety or effectiveness and does not put subjects at risk.
- A device intended solely for veterinary use.
- A device shipped solely for research on or with laboratory animals and labeled in accordance with 21 CFR 812.5(c) .
- A custom device, as defined in 21 CFR 812.3(b) , unless the device is being used to determine safety or effectiveness for commercial distribution.
- Custom devices are "limited to no more than 5 units per year of a particular device type" and require annual reporting to the FDA. For additional information, refer to the FDA Guidance for Industry and FDA Staff – Custom Device Exemption .
Exempted investigations for diagnostic devices
A diagnostic device is exempt from the IDE regulations, if the following four criteria accurately describe the use of the device in the planned clinical study. The device:
- (i) Is noninvasive,
- "A noninvasive device is one that does not, by design or intention:
- penetrate or pierce the skin or mucous membranes of the body, the ocular cavity, or the urethra; or
- enter the ear beyond the external auditory canal, the nose beyond the nares, the mouth beyond the pharynx, the anal canal beyond the rectum, or the vagina beyond the cervical os (21 CFR 812.3(k) )."
- (ii) Does not require an invasive sampling procedure that presents significant risk,
- While the device itself may be noninvasive, the sampling required to use a diagnostic device may be invasive, such as obtaining a biopsy. To determine if an invasive sampling procedure presents a significant risk:
- FDA recommends "…that you base your risk determination on the nature of the harm that may result from sampling. For example, FDA considers sampling techniques that require biopsy of a major organ, use of general anesthesia, or placement of a blood access line into an artery or large vein (subclavian, femoral, or iliac) to present a significant risk."
- "Blood sampling that involves simple venipuncture is considered noninvasive, and the use of surplus samples of body fluids or tissues that are left over from samples taken for noninvestigational purposes is also considered noninvasive (21 CFR 812.3(k) )."
- (iii) Does not by design or intention introduce energy into a subject, and
- Any form of energy qualifies (e.g. light, heat, X-ray, gamma ray, magnetic fields). The study can be exempt if energy is introduced as a part of clinical care and there is no additional energy introduced because of the study. For example, if the investigational diagnostic device is applied to X-ray images collected as a part of clinical care, then the study can be exempt.
- (iv) Is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure.
- "…test results …should not influence patient treatment or clinical management decisions before the diagnosis is established by a medically established product or procedure"
- "…If an investigational test uses a new technology or represents a significant technological advance, established diagnostic products or procedures may not be adequate to confirm the diagnosis provided by the investigational IVD."
- If the diagnostic is not being used to make treatment decisions for subjects in the study, then confirmation of the diagnosis is not required as a part of that study in order to meet this criterion.
For additional information about IVDs and the diagnostic device exemption criteria, please see the FDA Guidance for Industry and FDA Staff - In Vitro Diagnostic (IVD) Device Studies - Frequently Asked Questions .
What if my device study is not exempt from the IDE regulations?
- The study requires an IDE (full or abbreviated). However, in order to decide which type of IDE is needed, an SR/NSR determination is required.
- A non-significant risk (NSR) study requires an Abbreviated IDE and is solely overseen by an IRB.
- A significant risk (SR) study requires an IDE that is reviewed by the FDA.
What is an Abbreviated IDE?
- Oversight is by the IRB. FDA does not have direct oversight.
- Must meet the abbreviated IDE Requirements (21 CFR 812.2(b)(1) ):
- Label the device properly (21 CFR 812.5 )
- Obtain and maintain IRB approval after presenting why the device study is NSR
- Obtain informed consent (21 CFR 50 )
- Monitor the study (21 CFR 812.46 )
- Maintain required records and reports
- Comply with prohibitions against promotion (21 CFR 812.7 )
How is significant risk determined?
- The sponsor makes an initial risk assessment that is submitted to the IRB.
- The IRB then makes the significant risk determination, or if the IRB wants the FDA to make the risk assessment, a submission to FDA is necessary. For information on submitting a Study Risk Determination request to FDA go to the Study Risk Determinations from FDA page.
- A significant risk device is one that:
- Is intended as an implant and presents a potential for serious risk to the health, safety, and welfare of a subject.
- Is used to support or sustain human life.
- Is of substantial importance in diagnosing, curing, mitigating, or treating disease and/or otherwise preventing impairment of human health.
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.
- The study risk determination is based on the proposed use of a device in an investigation, and not on the device alone. (The same device can be in both an SR study and an NSR study.)
- If study is not SR, then it is NSR.
For additional information about significant risk determinations, please see the FDA Information Sheet Guidance For IRBs, Clinical Investigators, and Sponsors – Significant Risk and Nonsignificant Risk Medical Device Studies .